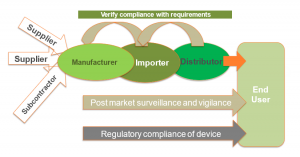
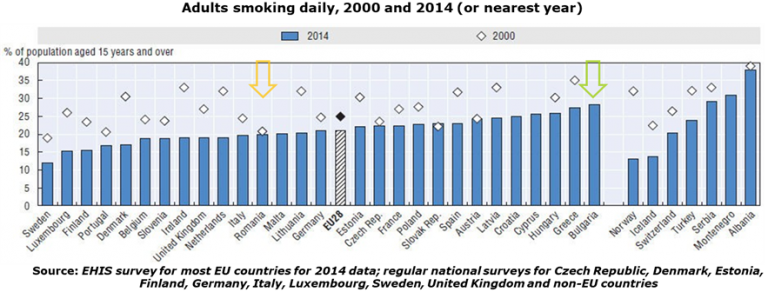
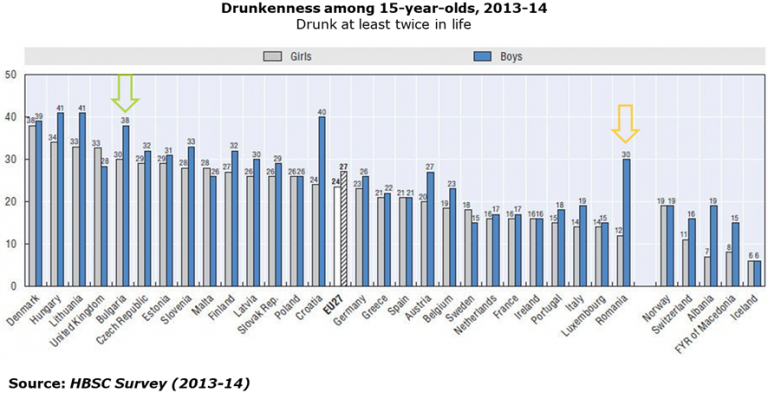
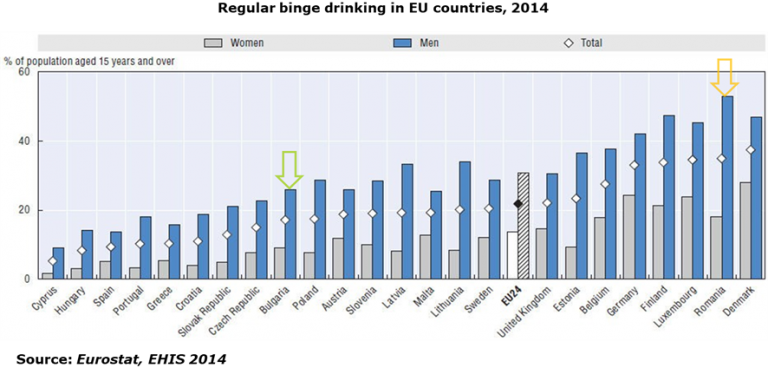
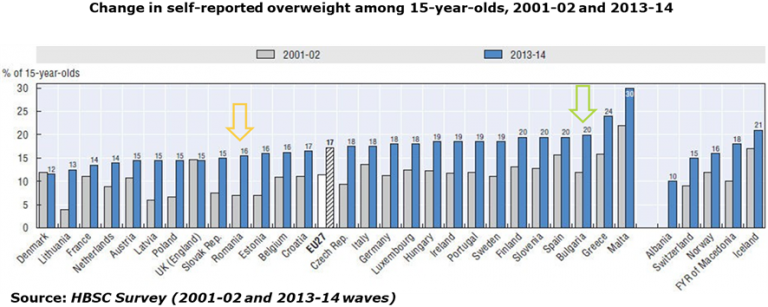
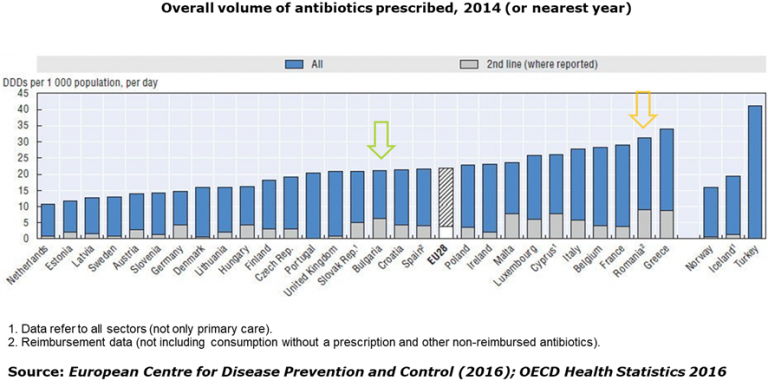
AICROS EXPERIENCE WITH COVID

There will be always patients requiring innovative treatment solutions, which at times may not be able to wait for COVID-19 to subside. For those cases we would like to see clinical research continuing as best as possible at a normal pace, what is not always easy, of course.
CROs are key in this process and we think our perspective will help to have a better picture of how COVID-19 is affecting and will affect Clinical studies.
The main challenges we have faced as CROs
In Ongoing trials:
We have lived in a very dynamic environment. There has been an increase in the use of online communication tools and an uptick in virtual team meeting to make sure that everyone is on the same page.
We were used to remote practices on operational tasks, but we have had to adapt all our business activities to a remote model.
Patient participation in the studies. The lack of patients was an issue for some trials and recruitment was directly on hold for many others.
It has been also a challenge for patients and our people to comply with COVID-19 regulations that, moreover, were constantly changing.
Responsiveness and availability of site staff has been rather limited for a lot of our trial sites, limiting traditional data verification options and delaying clinical trial progress. Most of this could be counteracted by increasing the amount of remote and centralized monitoring.
In those cases where onsite was preferable, like close out visits, travel to sites was subject to restrictions and had to be rescheduled.
Regarding new trials:
Fundraising for new research projects has been limited worldwide.
Many sponsors were hesitant to start new clinical trials, mainly because it was unclear how COVID-19 might impact ongoing trials.
Travel to sites has been limited for study start-up, site assessment and site initiation visits. Many of them have been cancelled or postponed due to lockdowns.
The information was changing so instruction from local regulatory bodies and sponsors has been unclear.

Some of the measures we have introduced in our procedures for running clinical trials in the context of the pandemic
Business Contingency Plans for clinical trials, that covered all facets of impacts that COVID-19 could have on clinical trials from availability of RA/EC for approval processes, to IMP/IMD shipments, availability of sites for on-site visits, how to handle patients that are on medication but cannot see the study site, to the need to adjust the statistical analysis or submit a protocol amendment to deal with the changes. It is very important that all this information is available for both our Project Management team and the Sponsor’s team.
Working with sites and clinics to implement virtual monitoring visits using electronic medical records and CRFs
CRA safety precautions, COVID-19 test before visiting a site.
Much more remote communication and increasing the number of remote visits.
Changes we have lived that are here to stay:
A lot of traditional monitoring techniques have not been conducted during the crisis, and remote and centralized monitoring techniques have played a much bigger role. On-site monitoring will be combined with remote monitoring as well as conducting virtual monitoring visits to minimize the cost of travel.
We remain hopeful that this change will persist, and that more and more authorities approve these procedures and more sponsors decide to move in this direction. Studies have shown that this results in improved data quality, and it can end up saving time and money that is otherwise spent on travel to the sites and performing Source Data Verification (SDV).
Additionally, we have seen home-sampling of laboratory samples employed – which can greatly improve the satisfaction of the patient. However, it may only be useful in niche trials and will likely remain irrelevant for the majority of clinical trials. Lastly, we have lived a more frequent use of e-signatures, opened up conversations with Regulatory bodies and e-Consent.
Digitalization of most of our activities was being completed before this situation started, and now it has been accelerated and we are pretty sure that, in a very near future, complete e-trials will be developed and we will find that results will be equally valid and patients will benefit from the results of that research.
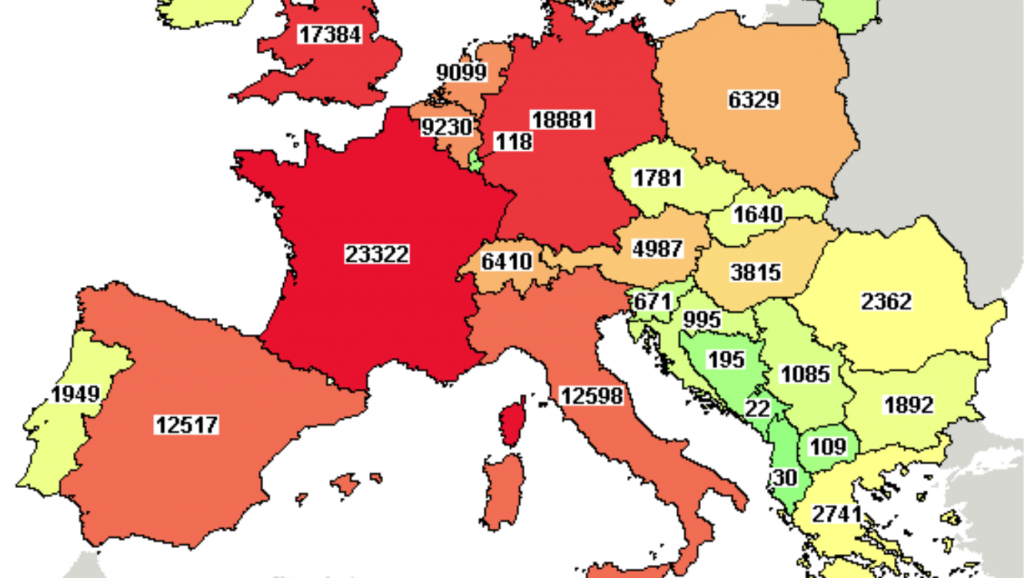
Our experience with COVID studies
At the moment there are almost 4.000 studies related to COVID-19 on clinicaltrial.gov database and as CROs we have had the opportunity to be part of some of them.
If you want to know more about global clinical investigation on Coronavirus and its evolution , you can read our in depth analysis that is updated every month in our Coronavirus research post.
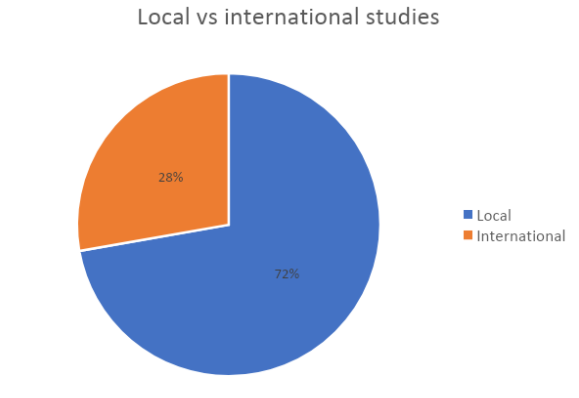
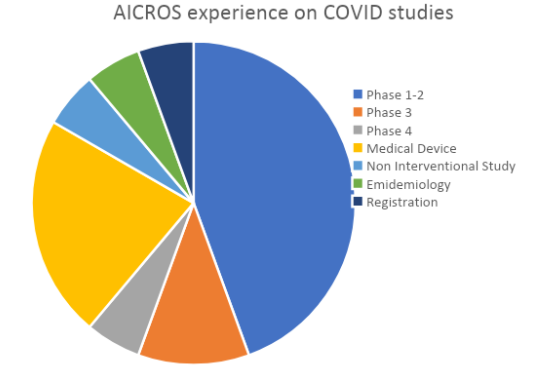
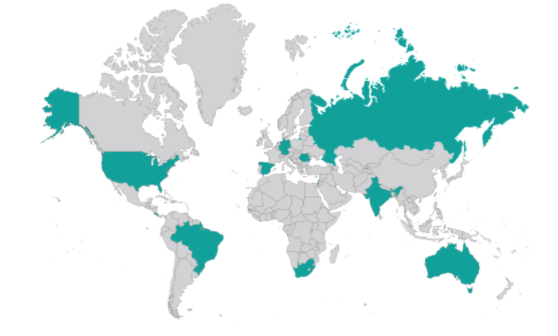
We highlight also that they have been conducted in a very wide range of geographic areas.
Although we have made use of the shortened regulatory paths that have been put in place by the regulatory authorities of many countries, we have so far seen limited impact on the studies we tried to conduct, and other trials going into the submission process have not been impacted. You can see more about this in this post.
How do we see the future of clinical trials?
We have all heard that crisis can also bring opportunities. In this case, COVID has helped us to glimpse possible trends in the future of outsourcing clinical trials.
Global clinical trials
COVID-19 has shed a light on the need to centralize clinical trials throughout the world and share this information to work together in order to advance together.
Remote monitoring
The coronavirus crisis has just speed up the trend and on-site monitoring will be reduced in the future
Patients’ visits remotely
Much more reliability on technologies that make people open to run not only monitoring but also Patients’ visits remotely.
Digital solutions on Monitoring
The use of electronic solutions not only on traditional activities such as Source Data Verification (SDV), but also Trial Master File (TMF), Site management, Investigator Site File (ISF) and even the consent procedure and Patient Reported Outcome (PRO) have shifted into focus for potential available electronic solutions.
Some of them can be seen as rather wide-spread but there is a lot of room for growth and development in the clinical research world.
We strongly believe that this is the right path to take, and that COVID-19 has accelerated change in this direction by forcing Sponsors, Sites and CROs alike to adapt.